
该研究于2016年发表于微生态领域期刊《The ISME Journal》上(影响因子:9.328)。
研究背景
过去十多年中,随着短读长、高通量二代测序的兴起,以16S rRNA基因部分可变区序列为目标的分析方法,逐渐替代了基于rRNA基因全长克隆文库的一代Sanger测序法,使我们能对菌群组成进行深度定量解析。然而,由于二代测序读长短的特性,我们无法对测得物种进行精确的分类鉴定,从而限制了我们对菌群代谢功能的深入理解。随着三代SMRT测序技术的出现,我们有望从根本上解决这一瓶颈,实现对rRNA基因全长序列的高通量精准测序。
研究方法
样本来源:23种细菌和3种古菌组成的人工菌群,以及取自湖水的天然微生物群落样本。
测序平台:PacBio RS II(16S rRNA基因全长)+Illumina MiSeq(16S rRNA基因V4区)。
对两种测序平台的测序结果进行多样性组成谱分析,并通过计算机模拟,比较16S rRNA基因全长序列和单V区序列的物种分辨能力。
研究结果
根据两种平台的测序结果,门水平的菌群组成较为相似,但在更精细的水平存在差异。同时,三代测序的结果显著降低了物种分类信息的不确定性。计算机模拟的结果也表明,短读长的单V区测序可能严重低估某些特定类群的微生物,比如水体样本中参与氮循环和甲烷代谢的物种。因此,基于三代测序的菌群多样性组成谱分析能极大地提升物种分类鉴定的精确性,实现“高分辨率”检测的同时,也为深入阐释菌群的代谢功能奠定了基础。
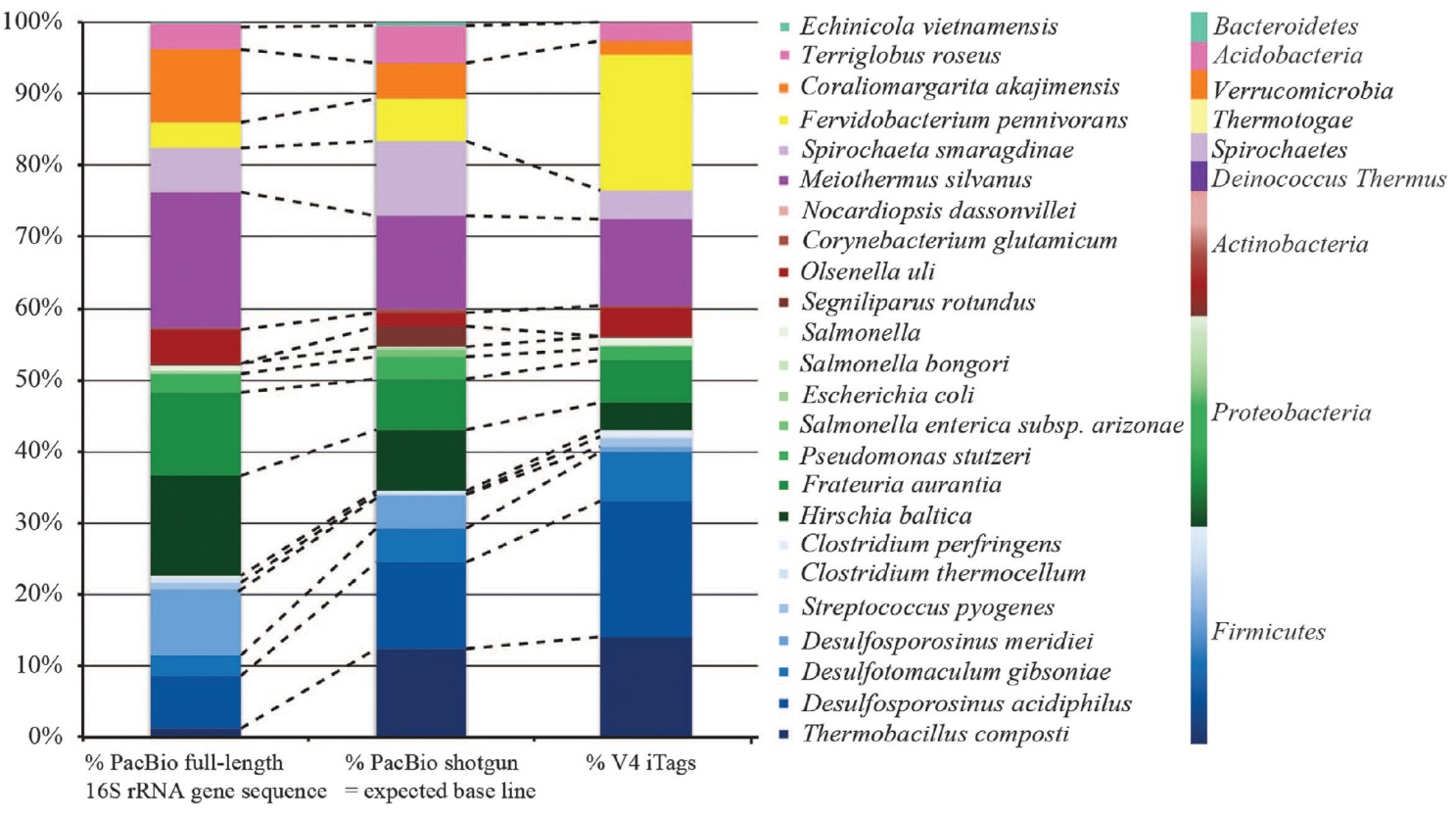
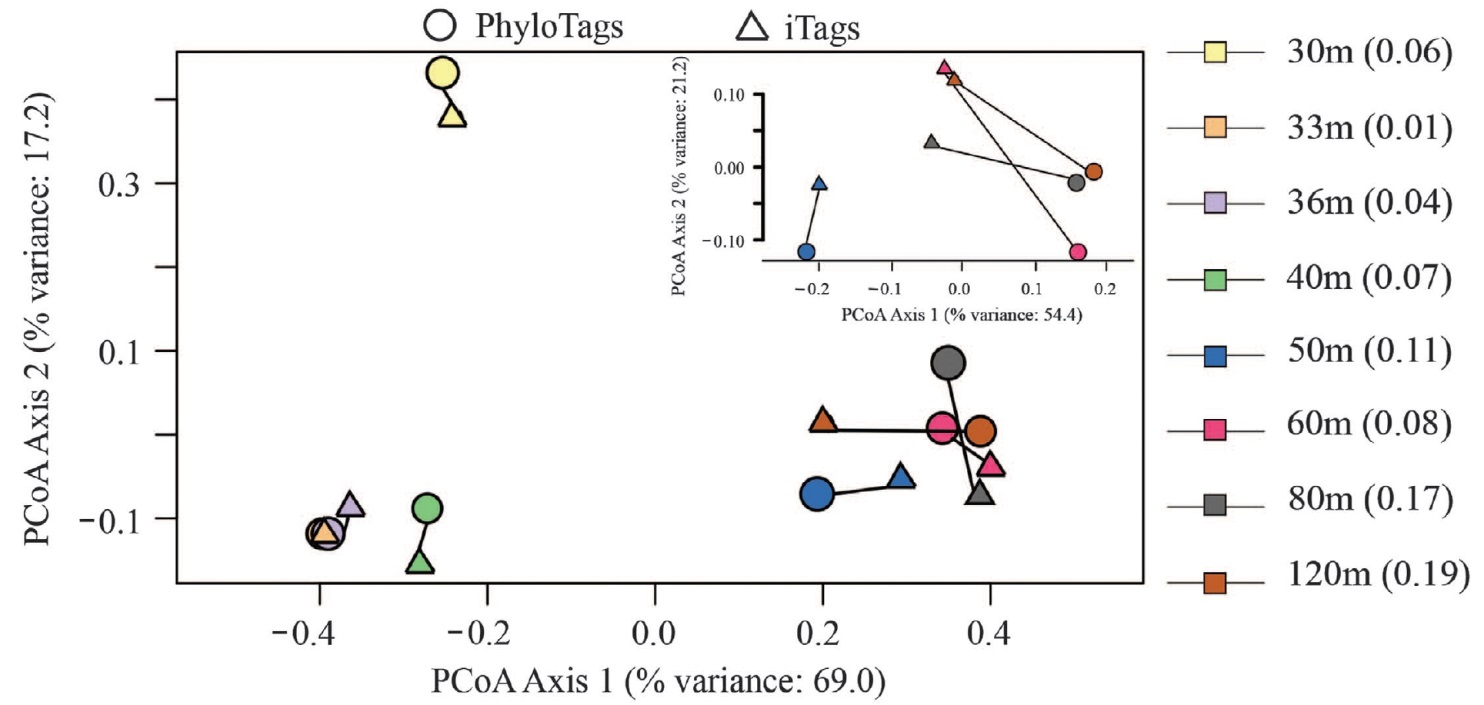
两种测序平台测序结果的整体比较。两者菌群的整体结构较为相似,但当菌群复杂程度增加时(底图子图),两者的检测结果有所差异。PhyloTags,三代PacBio RS II测序结果;iTags,Illumina MiSeq测序结果。
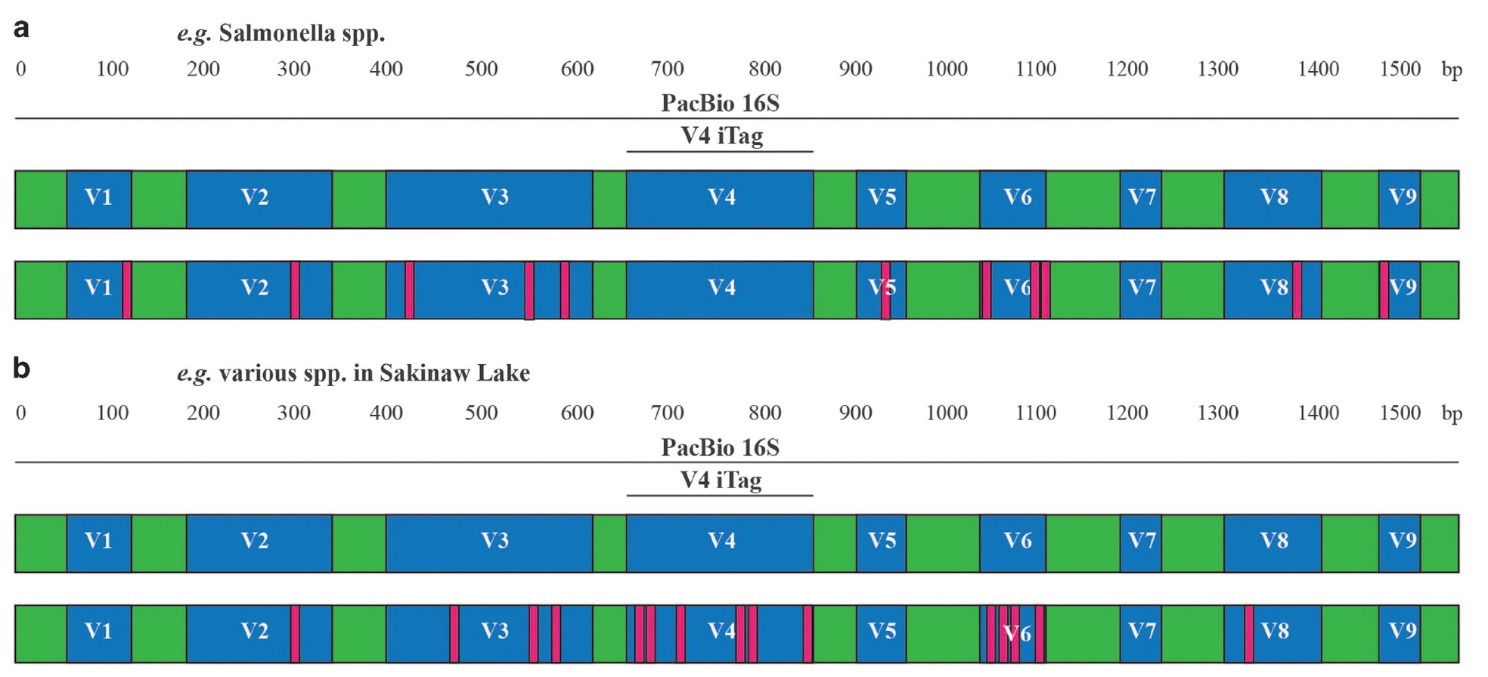
16S rRNA基因各V区的序列保守性并不一致,因而基于16S rRNA基因全长序列的三代测序,可以更全面地反映物种的种属信息。a图,以Salmonella属为代表,全长序列的种间差异达到97.4%,但V4区高度保守,二代测序结果将低估该物种的多样性;b图,某些物种在V4区的多样性可能高于其他V区,因而二代测序结果将高估相关物种的多样性。
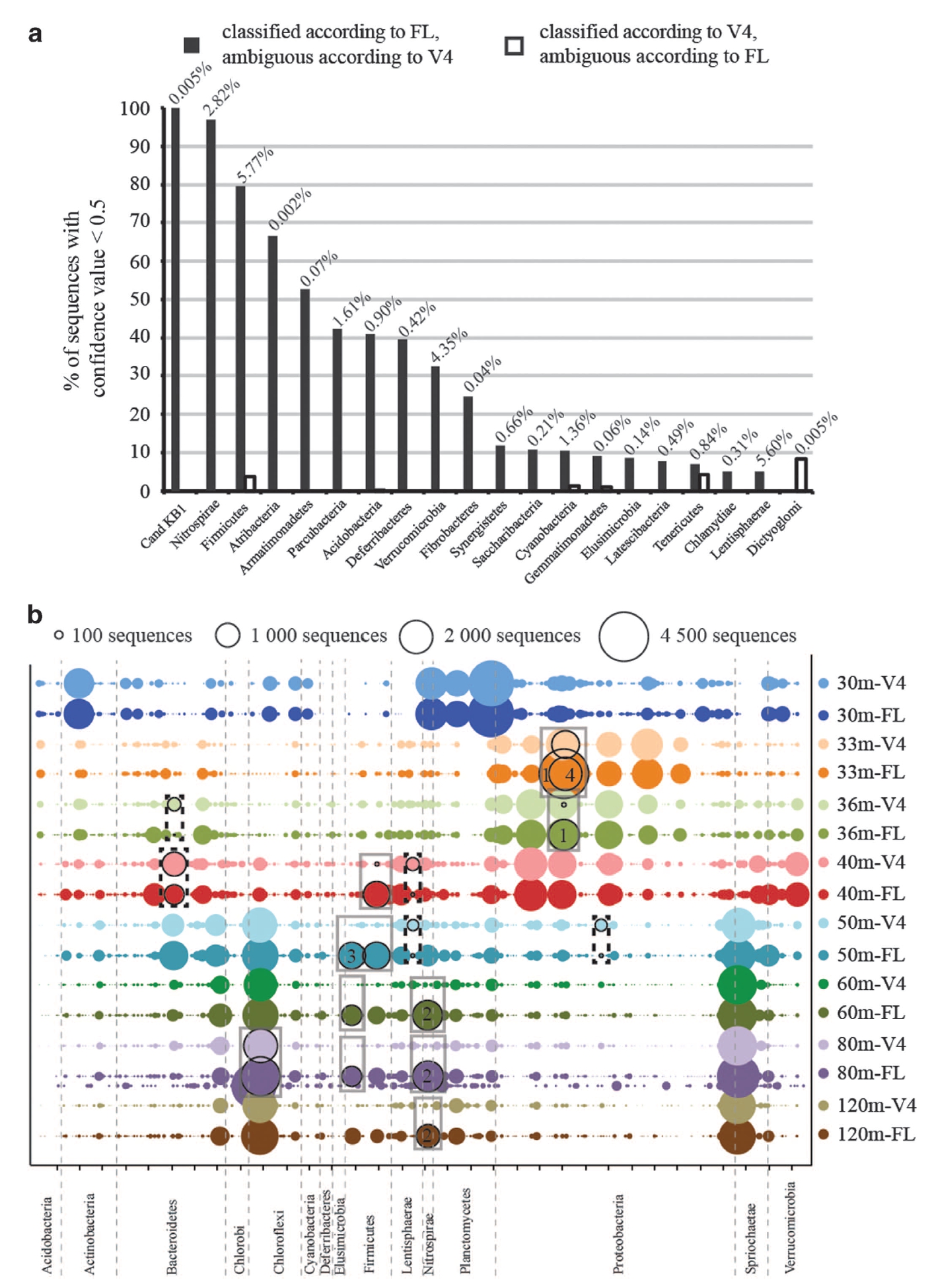
两种测序平台测序结果的精细比较。a图,三代测序的结果显著降低了物种分类信息的不确定性;b图,二代单V区测序结果可能会严重低估某些特定微生物类群的含量(以方框标记)。
研究结论
综上所述,运用三代测序技术进行微生物组研究,将显著提升菌群多样性组成谱解析的精准性和全面性。
参考文献
Singer, E., Bushnell, B., Coleman-Derr, D., Bowman, B., Bowers, R.M., Levy, A., Gies, E.A., Cheng, J.-F., Copeland, A., Klenk, H.-P., et al. (2016). High-resolution phylogenetic microbial community profiling. ISME J 10, 2020-2032.
